MDR-Ampel steht auf Gelb

Durch die Verschiebung der Umsetzungsfrist um ein Jahr auf den 26. Mai 2021 war in der Branche zunächst viel Erleichterung spürbar. Doch die Probleme wurden nur aufgeschoben und teilweise stehe der Markt noch da, wo er 2017 war, als die MDR in Kraft getreten ist. Dr. Bassil Akra kennt die MDR und den Markt seit Jahren, u. a. aus seiner Tätigkeit beim TÜV Süd. Von der Benannten Stelle ist er als Geschäftsführer zum Beratungsunternehmen Qunique GmbH mit Standorten in der Schweiz und Deutschland gewechselt.
Der benannte Flaschenhals
Und eben der Faktor der Benannten Stellen werde sich zum Flaschenhals in Sachen Umsetzung der MDR entwickeln. Sind für die Vorgänger-Vorschriften MDD noch über 50 Stellen benannt, wurden bis Mitte Februar gerade 19 für die MDR akkreditiert. Spezialgebiete, wie z. B. Implantate, decken nur Einzelne ab. Verschoben wurden infolge der Pandemie nicht nur die MDR-Frist um ein Jahr, sondern auch die fälligen Audits bei den Herstellern. Aufgrund von Kontakt- und Reisebeschränkungen konnten viele Benannte Stellen ihre Aufgaben nicht wahrnehmen. Und teilweise ist das auch heute noch so, weil zwar die Mitarbeiter inländischer Stellen zu den Herstellern reisen können, aber nicht die aus dem Ausland.

Fernaudits ermöglichen
Und von der EU ermöglichte Fernaudits seien eben nicht in allen Staaten erlaubt. So sollte die deutsche Regierung hierzu den Weg freimachen. Benannte Stellen seien bereit, dieses Verfahren anzuwenden, wenn es umsetzbar und praktikabel sei. Es könnte auf jeden Fall deren Arbeit erleichtern. Details sollten die Hersteller auch mit ihren Aufsichtsbehörden und Benannten Stellen klären.
MDD und MDR parallel
Außerdem laufen bei den Benannten Stellen derzeit die Zertifizierungen nach MDD und MDR gleichzeitig, was die Kapazitäten weiter begrenzt, so Dr. Akra. Weil viele Stellen noch keinen Antrag für eine Akkreditierung nach MDR gestellt haben, sollten die Hersteller bei ihren Benannten Stellen nachfragen, ob sie auch für die MDR tätig sein werden. Hersteller können bis Mai 2024 zudem gültige Zertifikate nach MDD als auch nach MDR haben. Aber auch wenn ein Unternehmen noch ein gültiges MDD-Zertifikat besitzt, müsse es die MDR-Vorgaben für die Marktüberwachung befolgen.
Engpass bei der EU
Eine weitere Engstelle machte der Referent auch bei der EU-Kommission aus. So suche auch die EU nach Spezialisten für eine für die MDR zuständige Expertengruppe, die im April/Mai ihre Arbeit aufnehmen könnte. Auch vonseiten der EU müssten in den nächsten drei Jahren zahlreiche Arbeiten erledigt werden. So gebe es mittlerweile zwar viele sog. Guidance-Papiere, allerdings ließen diese immer noch einen großen Interpretationsspielraum, so Dr. Akra. Außerdem fehlten noch viele Informationen vonseiten der EU für die Marktteilnehmer und Benannten Stellen. Daten für die jeweiligen Veröffentlichungen würden regelmäßig verschoben, z. B. zu den harmonisierten Standards.
Eile ist geboten
„Bitte warten“ beschreibt die Situation treffend. Allerdings sollten die Hersteller nicht länger zuwarten und die Hände in den Schoß legen, bis die Papiere alle vollständig vorhanden sind. Entsprechend sollten nach Rücksprache mit den Benannten Stellen die internen Abläufe und Dokumentationen nach den aktuellen Kenntnisständen vorbereitet werden, etwa indem geklärt wird, welche Aspekte der Technischen Dokumentation (TD) für eine fokussierte Prüfung herangezogen werden.
Dies betreffe auch die noch nicht vorhandenen EMDN-Codes, entsprechend einer noch nicht veröffentlichten europäischen Nomenklatur für die Produkte. Gleichzeitig sollten die Hersteller verfolgen, was es vonseiten der Kommission Neues gibt. Dr. Akras dringender Appell an die Hersteller: „Verlieren Sie keine Zeit!“

Fehler vermeiden
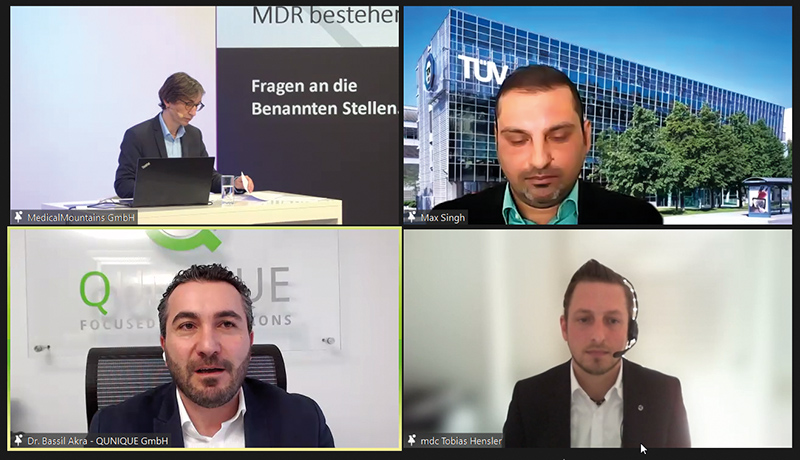
Die Sicht der Benannten Stellen brachten Tobias Hensler von der MDC Medical Device Certification GmbH und Dr. Max Singh von der TÜV Süd Product Service GmbH ein. Ihre Berichte aus der Praxis waren bei der Tagung mit dem plakativen Ziel und Wunsch zugleich verbunden: „MDR bestehen“. Den beiden Referenten ging es darum, Herstellern Tipps für die Vermeidung von Fehlern zu geben.
Hensler stellte MDC als akkreditierten Zertifizierer für QM-Systeme und Benannte Stelle nach MDR, MDD und IVDD vor. Insgesamt seien mehr als 90 Mitarbeiter angestellt sowie über 70 Auditoren und Fachexperten freiberuflich für die Firma tätig. Seit 2019 gibt es auch einen Standort in Tuttlingen neben Stuttgart, Berlin und Wien. MDC habe bereits acht Wochen nach der Benennung erste MDR-Verfahren durchführen können.
Die große Welle kommt noch
Auch Hensler verwies darauf, dass die MDR für die Benannten Stellen mehr Anforderungen und Formalismus bringe. Und die große Welle komme auf die Benannten Stellen erst noch zu. Denn während 2020 knapp 600 Zertifikate nach MDD und AIMD ausgelaufen sind, werden es heuer rund 1.550 sein. Und für 2022 rechnen die Benannten Stellen mit rund 1.850, 2023 über 2.700 und 2024 fast 7.300 auslaufenden Zertifikaten. Allein dieser Transfer von MDD zur MDR binde viele Kapazitäten bei den Stellen. Hensler verwies zudem darauf, dass bei der MDD die Prüfung der Technischen Dokumentation und das Audit selber noch parallel laufen können.
Bei der MDR müsse zunächst die Technische Dokumentation abschließend geprüft werden, bevor mit der Auditplanung begonnen werden kann. „Sollte es Abweichungen in der Technischen Dokumentation geben, verzögert sich alles“, betonte Hensler. Deshalb sollten die Hersteller sehr viel Wert auf eine die Vorgaben erfüllende Dokumentation legen. Aus der Praxis verwies er auf einige Erkenntnisse zur Prüfung der Produkte der Klasse Ir.
So sei immer wieder die Definition des wiederverwendbaren chirurgischen Instrumentes nicht eindeutig gewesen. Auch Zweckbestimmung und Anwendungsgebiet seien nicht ausreichend betrachtet worden. Das gelte auch für Beschichtungen. Es komme auch vor, dass Laborergebnisse nicht abschließend bewertet oder nicht richtig zugeordnet werden. Beim Aufbereitungszyklus sollten zudem die maximale Anzahl der Aufbereitungen und die Anforderungen an alle Phasen betrachtet werden – dies beinhaltet: Vorbereitung am Gebrauchsort, Vorbereitung der Reinigung und Desinfektion, Trocknen, Kontrolle – Wartung – Prüfung, Verpackung, Sterilisation und Lagerung.
Knackpunkt Technische Dokumentation
Auch im Abgleich von QM-System und MDR finden sich immer wieder Hürden. So komme es vor, dass die Validierungsergebnisse aus der Technischen Dokumentation nicht die Prozesse und Herstellschritte widerspiegeln. Wenn die Regelungen zwischen Lieferant und Hersteller nicht eindeutig sind, kann das zu Problemen führen; die Verantwortung dürfe nicht einfach an den Lieferanten abgegeben werden.
Außerdem fänden sich immer wieder die gleichen Abweichungen in Technischen Dokumentationen, weil die Ergebnisse aus den TD-Prüfungen nicht systematisch auf andere Dokumentationen übertragen werden. Hensler: „Die Dokumentenlenkung und Aktualisierung der Technischen Dokumentation findet nicht in ausreichendem Maße statt.“ Immer wieder stelle man als Benannte Stelle fest, dass die Kunden den aktuellen Stand der technischen Normen nicht verwenden. Leichtsinnsfehler bestünden auch darin, bei der Konformitätserklärung oder Gebrauchsanweisung auf die alte Richtlinie zu referenzieren.
Und selbst wenn die europäische Datenbank Eudamed noch nicht läuft, sollten die Hersteller das Verfahren zur eindeutigen Produktidentifikation UDI für die einzelnen Produkte festlegen.

Häufige Fehlerquellen
Erfahrungen aus der Praxis teilte auch Dr. Max Singh, der beim TÜV Süd globaler Leiter des Orthopedic-Focus-Teams ist. Er verfügt über rund 15 Jahre Erfahrung aus Industrieunternehmen, hat u. a. auch in Tuttlingen gearbeitet. Wie er darlegte, können Hersteller entscheiden, ob sie Produkte zunächst noch weiterhin unter der bisherigen MDD in den Markt bringen oder sie zur MDR transferieren. Hersteller sollten mit ihrer Benannten Stelle klären, ob diese alle Produktgruppen aus dem Sortiment abdecken kann. Mit Stand Anfang Februar konnten z. B. nur 4 der 18 Benannten Stellen eine breitere Palette abdecken, darunter nur eine aus Deutschland. Auch er verwies auf die ersten Erfahrungen mit Prüfungen der Technischen Dokumentationen und häufigen Fehlerquellen. Die meisten Abweichungen gab es demnach in den Bereichen:
- Risiko-Management
- Gebrauchstauglichkeit
- Biokompatibilität
- Design & Herstellungsprozesse
Teilweise seien die strukturellen Vorgaben für Technische Dokumentationen nicht eingehalten worden. Dr. Singh: „Umso besser die Technische Dokumentation organisiert und strukturiert ist, umso schneller kommt die Benannte Stelle zu einem Resultat.“ In der TD sollten deshalb Zusammenfassungen vorangestellt werden. Auch die Marktüberwachungs-Aktivitäten würden nicht ausreichend beschrieben. Beim Risikomanagement seien z. B. Zweckbestimmung und Kontraindikationen nicht ausreichend dargestellt.
Beim Punkt Design und Herstellung fehlen teilweise die Bewertung der Toxikologie, Materialspezifikationen und Schnittstellen oder auch eine detaillierte Beschreibung des Herstellungs- und Designprozesses auf Produktebene. Zudem sei es wichtig, die in der Produktion verwendeten Hilfsmittel zu erwähnen. Auch Bewertungen zur Gebrauchstauglichkeit oder zur Verpackung seien oft nicht vollständig. Fehlerquellen seien in Gebrauchsanweisungen und unvollständigen Kontaktdaten zu finden. Auch seien Informationen zum Produkt im Internet nicht vollständig oder nicht aktuell. Nicht fehlen dürfen klinisch relevante Dokumente, Verweise auf die vorigen Generationen des Produktes sowie die Lebensläufe wichtiger Experten aus dem Team.
- Dr. Singh gab den Teilnehmern noch folgende Hausaufgaben mit auf den Weg:
- Die regulatorische Abteilung personell stärken.
- Im Gespräch mit der Benannten Stelle bleiben und mit ihr die MDR-Zulassung planen.
- Informiert bleiben über die aktuellen Entwicklungen und Dokumente.
- Nicht länger warten mit der MDR-Umsetzung.
Audits bei Karl Storz und Richard Wolf
Von ihren Erfahrungen mit den MDR-Audits durch ihre Benannten Stellen berichteten Karim Djamshidi von Karl Storz und Wulf Brunow von Richard Wolf. Bei Storz liegen die MDR-Zertifikate bereits für viele Klassen vor, bei Richard Wolf zunächst für Klasse IIa. Beide verwiesen darauf, dass die Audits sehr theoretisch abliefen, aber vergleichbar mit anderen Audits seien. Bei Storz habe man gleich mehr Zeit für die Auditierung eingeplant. Außerdem seien zwischen dem Audit und dem Erhalt des Zertifikats rund neun Monate vergangen.
Interner Testlauf
Djamshidi gab den Herstellern den Rat, bereits intern ein Audit zur MDR zu durchlaufen, an diesem Prozess könnten die Benannten Stellen sich dann entlanghangeln. Zu den praktischen Tipps zählen auch, für Haftpflichtversicherungen ausreichende Deckungssummen zu berechnen. Zudem werde die Fähigkeit für Übersetzungen in die Amtssprachen der EU kontrolliert, sofern in die betreffenden Länder exportiert werde. Brunow verwies darauf, dass die Übersetzung mit externen Partnern auch vertraglich geregelt werden sollte. Gerade die Struktur der Technischen Dokumentation unterscheide sich bei der MDR gegenüber der vorherigen MDD.
Er empfahl, für die intern jeweils verantwortlichen Führungskräfte auch Stellvertreter zu benennen und die jeweiligen Schulungsnachweise zu dokumentieren. Dazu gehört auch, so ergänzte Djamshidi, intern klare Berichtsstrukturen aufzubauen und diese auch umzusetzen. Einzelne Detail- oder Verständnisfragen könne man im Vorfeld mit Benannten Stellen klären. Diese dürften jedoch keine ausführliche Beratung anbieten. Hier sollte man deshalb ggf. kompetente Hilfe von außen in Anspruch nehmen, so Brunow. Er riet, regelmäßig Projektbesprechungen mit der Benannten Stelle zu vereinbaren.
MDR als Anstoß zum Bereinigen des Portfolios
Beide verwiesen darauf, dass im Zuge der MDR das Produktportfolio auch bereinigt werde. Die MDR habe dazu den notwendigen Anstoß gegeben. Weil man so viele Verpflichtungen eingehe, müsse für jedes einzelne Produkt überprüft werden, ob sich der Aufwand noch lohne. Beide Firmenvertreter erklärten, dass die Anforderungen auch die Kapazitäten für Neuentwicklungen einschränken. In den USA könnten Produkte inzwischen rascher zugelassen werden als in Europa.
Ein anderes Thema ist der Umgang mit den Zulieferern und die Auswirkungen der MDR auf diese. So gebe es auch Zulieferer, die sich wegen der Anforderungen der MDR aus dem Medizintechnik-Sektor verabschiedet hätten. Djamshidi unterstrich, dass von den Zulieferern vollständige und ungeschwärzte Quellenunterlagen vorliegen müssen.